a cura di Giuseppa Lo Surdo1, Lorenzo Di Spazio2, Paolo Baldo3, Maria Cecilia Giron4.
- U.O.C. Farmacia Ospedaliera, Fondazione Toscana ‘Gabriele Monasterio’, Massa
- S.C. Farmacia Ospedaliera Nord, Ospedale S. Chiara di Trento, Azienda Provinciale per i Servizi Sanitari
- SOSD Farmacia Ospedaliera, Centro di Riferimento Oncologico CRO Aviano -IRCCS
- Dipartimento di Scienze del Farmaco, Università di Padova
Abstract
Il nuovo Regolamento Europeo sui Dispositivi Medici, MDR 2017/745, è pienamente in vigore dal 26 maggio 2021, dopo essere stato posticipato di 12 mesi a causa dell’emergenza pandemica. Il nuovo regolamento non stravolge drasticamente il quadro legislativo preesistente ma fornisce sicuramente una strategia armonizzata e appropriata per la gestione dei dispositivi medici, basata sul loro ciclo di vita. Rafforza l’approccio esistente in materia di supervisione degli “Organismi Notificati” e disciplina in maniera univoca le sperimentazioni precliniche e cliniche e la vigilanza-sorveglianza sul mercato. Da ultimo, ma elemento non meno importante, il nuovo regolamento pone l’accento su requisiti di qualità e principi di trasparenza e tracciabilità lungo tutta la filiera commerciale e il ciclo di vita dei dispositivi medici. Il risultato atteso è quello di garantire livelli elevati di qualità e sicurezza sostenendo allo stesso tempo l’innovazione tecnologica. Cosa dobbiamo aspettarci e quali saranno le possibili ripercussioni sul servizio di Farmacia Ospedaliera?
Last Update: 10/05/2021
Introduzione
Entrato in vigore il 25 maggio 2017, il nuovo Regolamento Europeo sui Dispositivi Medici (MDR)[1] abroga le direttive 93/42/EEC (Medical Devices Directive- MDD) e 90/385/EEC (Active Implantable Medical Devices Directive- AIMDD) e cerca di rispondere alle necessità normative derivanti dagli importanti progressi tecnologici compiuti nell’arco dell’ultimo ventennio (ad esempio relativamente a Dispositivi Medici (DM) estremamente innovativi e/o con tecnologie ibride).
Questo regolamento, molto atteso, intende fornire un quadro normativo solido, trasparente e sostenibile, riconosciuto a livello internazionale, e si pone come obiettivo quello di migliorare la sicurezza clinica e creare un accesso equo al mercato per i produttori.
Contrariamente alle direttive, che devono essere recepite nel diritto nazionale, i regolamenti hanno portata generale, sono obbligatori in tutti i loro elementi e direttamente applicabili negli ordinamenti degli Stati membri. Pertanto l’applicazione del regolamento appianerà le divergenze tra sistemi regolatori nazionali e ridurrà significativamente i rischi di discrepanze nell’interpretazione nel mercato dell’Unione Europea (UE).
Aspetto molto importante, il nuovo quadro normativo introduce un approccio life-cycle in merito al rapporto efficacia/sicurezza del DM, potenziando la necessità di raccolta dati relativi ad efficacia e sicurezza, la sorveglianza post-commercializzazione e garantendo un livello elevato di tracciabilità del dispositivo.
Timeline del MDR [2, 3]
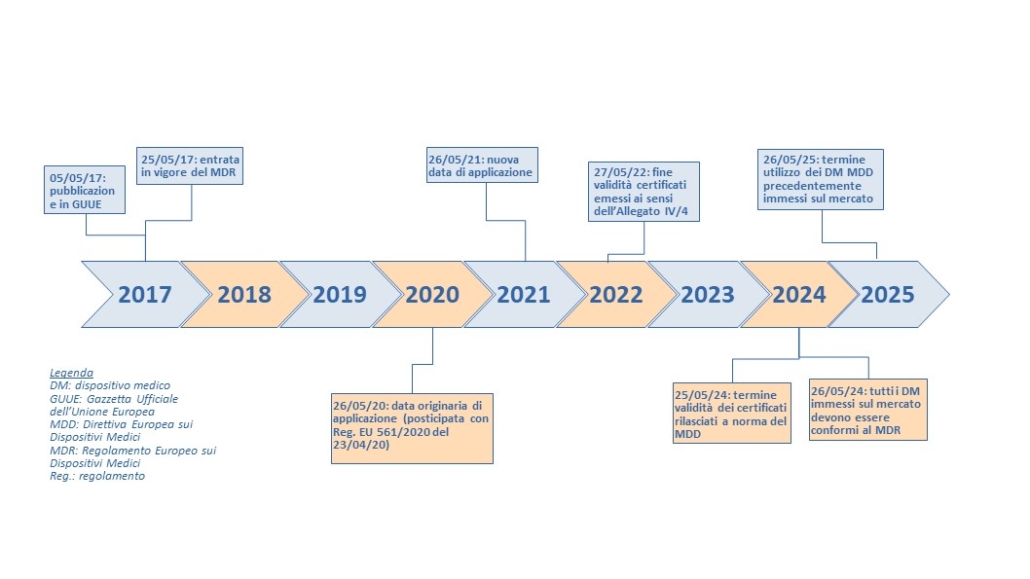
Cosa cambia:
- campo di applicazione più ampio (Articoli 1-2, Allegato XVI). Con l’applicazione del nuovo MDR vengono definiti come DM, ad esempio, anche tutti i dispositivi per la pulizia, la sterilizzazione o la disinfezione di altri DM, i dispositivi sottoposti a ricondizionamento e i dispositivi che non hanno una specifica destinazione d’uso in ambito medicale (es. lenti a contatto colorate);
- definizione precisa degli Operatori Economici (OE) coinvolti nella catena di fornitura dei DM (Fabbricante, Mandatario, Importatore e Distributore) (Articolo 2.30 e 2.32-34); viene fornita una descrizione dettagliata delle responsabilità e degli specifici adempimenti obbligatori per ciascuna figura (Articoli 10-14);
- obbligo di individuare una Persona Qualificata per i produttori di DM, responsabile della verifica della conformità ai requisiti del nuovo MDR (Articolo 15);
- criteri stringenti per la designazione degli Organismi Notificati (ON; Articoli 35-50) e più ampi poteri (fungeranno da esecutori della nuova normativa), compreso il diritto di effettuare audit senza preavviso e test continui sui DM per garantirne la conformità anche dopo l’ottenimento della certificazione;
- nuove regole per determinare la classe di rischio del DM (Articolo 51, Allegato VIII); sono stati identificati requisiti molto più specifici sulla base del rischio a cui viene esposto il paziente che comporterà un aumento di classe per molti dispositivi;
- la CND verrà adottata a livello europeo (European Medical Device Nomenclature-EMDN; Articolo 26);
- introduzione di una procedura coordinata per la valutazione delle indagini cliniche condotte in più di uno Stato membro;
- una più ampia documentazione tecnica di valutazione della sicurezza preclinica da produrre; l’Allegato I specifica i requisiti generali di sicurezza e prestazione, mentre gli Allegati II e III delineano la struttura della documentazione tecnica;
- introduzione di un sistema di tracciabilità basato sull’Unique Device Identification (UDI) (Articoli 25 e 27, Allegato VI parte C);
- nuovi requisiti per etichetta e IFU (Allegato I);
- la predisposizione di una card per paziente per i dispositivi impiantabili (Articolo 18);
- evidenza di una maggiore trasparenza, per gli operatori sanitari e per il pubblico, assicurata grazie all’istituzione di una banca dati europea multimodale completa e pubblica sui DM (European Database on Medical Devices, EUDAMED; Articoli 28-34); EUDAMED faciliterà il coordinamento tra gli Stati membri, permettendo di razionalizzare e facilitare il flusso di informazioni tra tutti gli attori coinvolti. Questo database presenterà 6 moduli interconnessi:
- Registrazione degli OE;
- Registrazione UDI/dispositivi;
- ON e Certificati;
- Indagini cliniche;
- Vigilanza e sorveglianza post-commercializzazione;
- Sorveglianza del mercato,
Il rilascio di EUDAMED avverrà in 3 fasi: il primo modulo sulla registrazione degli attori è stato reso disponibile da dicembre 2020. Mentre i moduli per la registrazione dei singoli DM e degli ON sono disponibili da maggio 2021. Per i restanti moduli non sono ancora disponibili previsioni sulla data di rilascio[4] ma comunque un primo rilascio “go alive” dovrebbe essere previsto per maggio 2022;
- vigilanza e sorveglianza post-market più rigorose (Articoli 83-90). I fabbricanti dovranno raccogliere e analizzare i dati clinici post-market come parte della valutazione continua del profilo di sicurezza ed efficacia, e rendere disponibile relativo rapporto di sorveglianza (per i dispositivi di classe I) o un rapporto periodico sulla sicurezza (per i dispositivi di classe IIa, IIb e III).
- per la dispositivo-vigilanza viene introdotta una modifica della definizione di “incidente” (Articolo 2 comma 64) e introduzione di “incidente grave” (Articolo 2 comma 65) e “grave minaccia per la salute” (Articolo 2 comma 66). Inoltre, vengono assegnati precisi ruoli e obblighi, non solo ai fabbricanti e mandatari, ma anche agli importatori (Articolo 13 commi 8 e 10) e ai distributori (Articolo 14 comma 5).
Inoltre, viene introdotto un rafforzamento delle norme sulle evidenze cliniche (Articolo 61-62 e Allegato XV) da produrre per l’ottenimento della marcatura CE; il contenuto di un intero paragrafo e 2 allegati (capo VI, Articoli 61-82 e Allegati XIV-XV) intende disciplinare la la valutazione clinica e le indagini cliniche da condurre utilizzando DM. A differenza del MDD, viene introdotto il principio di equivalenza secondo il quale è possibile dimostrare l’equivalenza di un nuovo dispositivo ad un altro già esistente se è stato stipulato un contratto tra i due fabbricanti secondo quanto stabilito dall’art. 61 comma 5 e l’equivalenza deve essere secondo quanto indicato nell’Allegato XIV comma 3. Inoltre, i DM immessi sul mercato prima dell’applicazione del nuovo regolamento (anche se non riclassificati) richiederanno comunque la rivalutazione dei dati clinici.
Cosa aspettarsi e quali opportunità
Nell’aprile 2019, l’associazione europea che riunisce le aziende del settore dei presidi bio-medicali ‘MedTech Europe’ ha reso pubblica una lettera aperta diretta alla Commissione Europea[5]. Secondo tale documento, la principale preoccupazione sarà associata alla drastica riduzione del numero di ON per la valutazione della conformità dei DM. Ad oggi da Database europeo NANDO risultano accreditati solo 20 organismi, di cui appena 2 in Italia[6].
Poiché tutti o quasi i dispositivi richiederanno una nuova certificazione da parte di un ON, il numero ridotto di tali enti potrebbe comportare tempi più lunghi per l’immissione in commercio (time to market) e costi maggiori per lo sviluppo di nuovi DM per i fabbricanti. Sicuramente, in un primo momento l’obiettivo principale dei fabbricanti sarà rappresentato dalla ri-certificazione dei dispositivi già esistenti con possibile ritardo nello sviluppo di nuove tecnologie innovative. La necessità di dover produrre evidenze cliniche inoltre inciderà anche sull’attività dei comitati etici.
Il Regolamento sicuramente alza il livello di attenzione non solo sulla sicurezza dei dispositivi, ma anche sulla loro efficacia, conferendo formalmente a tutti gli OE importanti responsabilità di controllo sulla produzione e sulla commercializzazione del dispositivo. Ciò significa che per poter dimostrare la conformità dei propri dispositivi al Regolamento, e al contempo per verificarne in modo proattivo il rispetto dei requisiti di sicurezza ed efficacia, i fabbricanti sono tenuti ad istituire e mantenere un sistema idoneo per raccogliere ed analizzare dati provenienti “dal campo” relativi all’effettivo utilizzo degli stessi. Varie fonti di raccolta dei dati possono essere sfruttate ma sicuramente la più efficace è quella cosiddetta “proattiva” cioè coinvolgere gli utilizzatori richiedendo loro un giudizio diretto su vari aspetti che riguardano le prestazioni, la sicurezza e l’usabilità dei DMi.
Oltre ad ottenere un innalzamento dei livelli di sicurezza e qualità nel campo dei DM, il nuovo Regolamento crea, quindi, un terreno fertile per la nascita di nuove partnership scientifiche tra ricercatori, operatori sanitari, clinici e fabbricanti. A tal proposito sarebbe sicuramente utile che, a livello europeo o nazionale, venissero definite delle linee guida condivise che diano spazio e permettano di strutturare percorsi di collaborazione virtuosi tra istituzioni sanitarie e aziende.
Infine, nonostante gli obblighi prescritti dal MDR ricadano principalmente su OE e organismi competenti, anche le strutture del sistema sanitario sono chiamate ad incrementare le attività legate alla sorveglianza e vigilanza del mercato dei DM grazie soprattutto al sistema UDI. A tal proposito l’Articolo 27, al comma 9, obbliga le istituzioni sanitarie a registrare e conservare, di preferenza per via elettronica, gli UDI dei DM appartenenti alla classe III che hanno ricevuto. Per quanto riguarda i dispositivi appartenenti alle classi di rischio inferiore, gli stati membri hanno l’onere di ‘incoraggiare’ le istituzioni sanitarie, e possono, addirittura, obbligarle, a registrare e conservare, di preferenza per via elettronica, gli UDI dei dispositivi che hanno ricevuto. Pertanto, per allinearsi a tali raccomandazioni, le strutture del SSN dovranno programmare, se non già presenti, l’implementazione di sistemi informatici per la gestione e tracciabilità dei DM impiantabili e non, con possibili vantaggi non solo in ambito di dispositivo-vigilanza ma anche a livello gestionale, come ad esempio la semplificazione del controllo delle scorte e scadenze e della rendicontazione clinica, l’automatizzazione dei controllo sui consumi, riordini e limiti di budget, a supporto del governo clinico e della gestione del rischio clinico.
Il nuovo MDR potrebbe avere un ruolo chiave nel promuovere e implementare la governance dei DM sia a livello europeo che a livello locale. L’obbligo, in particolare, di fornire evidenze precliniche e cliniche, al fine di ottenere la marcatura CE, potrebbe consentire di avere a disposizione un maggior numero di dati di efficacia e sicurezza per una valutazione omogenea e temporale del beneficio terapeutico determinato dal DM. Inoltre, l’introduzione di un sistema di tracciabilità dei DM univoco a livello europeo potrà favorire e potenziare la raccolta di dati di consumo, di durabilità e di eventi avversi, e di conseguenza rafforzare la sicurezza dei DM. La raccolta sistematica dei dati e l’analisi delle evidenze scientifiche permetteranno di confrontare il DM con i suoi equivalenti e/o comparatori e di determinare differenze clinicamente rilevanti, generando così un percorso di cura efficiente. Di conseguenza, in un’ottica di ottimizzazione delle risorse e della spesa sanitaria, questi dati potranno essere utilizzati per migliorare la capacità di governare l’innovazione tecnologica ed il progresso clinico-scientifico, assicurando un’efficiente allocazione delle risorse.
Bibliografia
- Regolamento (UE) 2017/745 del Parlamento europeo e del Consiglio del 5 aprile 2017 relativo ai dispositivi medici. Gazzetta Ufficiale dell’Unione Europea L 117/1, 5 maggio 2017
- Rettifica del regolamento (UE) 2017/745 del Parlamento Europeo e del Consiglio, del 5 aprile 2017, relativo ai dispositivi medici, che modifica la direttiva 2001/83/CE, il regolamento (CE) n. 178/2002 e il regolamento (CE) n. 1223/2009 e che abroga le direttive 90/385/CEE e 93/42/ CEE del Consiglio. Gazzetta Ufficiale dell’Unione Europea L 334/165, 27 dicembre 2019
- European Commission, 2019. Transition Timelines from the Directives to the Regulations Medical Devices and in vitro Diagnostic Medical Devices. Disponibile al sito: https://ec.europa.eu/docsroom/documents/34907. Accesso verificato il 26 maggio 2021
- Medical Devices – EUDAMED. Disponibile al sito: https://ec.europa.eu/health/md_eudamed/overview_it. Accesso verificato il 10 maggio 2021
- Bernasconi, S. Open letter on the implementation and readiness status of the new Medical Device Regulation 745/2017 (MDR). MedTech Europe, 2019. Disponibile al sito: https://www.medtecheurope.org/wp-content/uploads/2019/04/MedTech-Europe_VP-Katainen_MDR-implementation-status_15-April-2019.pdf. Accesso verificato il 26 maggio 2021
- European Commission, Database NANDO. List of Bodies Notified under directive: Regulation (EU) 2017/745 on medical devices. Disponibile al sito: https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34). Accesso verificato il 10 maggio 2021.